Using LINQ for Computational Genomics
I’ve been playing around a bit lately with computational genomics (I’m doing a project for my parallel computation class). I wanted to write some simple algorithms that operate on potentially large amounts of DNA data without using a ton of RAM. For example, the entire human genome is 3 billion base pairs – reading it all into memory is out of the question (at least on my home PC). It occurred to me that this was a perfect opportunity to spend some more time using LINQ. Computations like this are inherently stream based, and LINQ allows you to express and compose operations on streams very effectively. Here’s a simple example that I hope will help demonstrate the power of LINQ.
One of the simplest forms of statistical analysis done on a DNA sequence is producing a graph of it’s “GC density”. To a computer scientist, a DNA sequence is just a string of A, C, T, and G characters. The GC density with window size ‘w’ for a sequence is another (similarly sized) sequence of numbers between 0 and 1, each of which represents the ratio of Gs and Cs in the previous ‘w’ characters. For example, for the sequence “ATGCAG”, the GC density with window size 2 is “0, 0.5, 1, 0.5, 0.5”.
So, in order to calculate this, I first started with a simple function which, given a nucleotide sequence, would return an identical-length sequence of the number of Gs or Cs seen so far:
/// <summary>
/// Generate a sequence that is the prefix sum of Gs and Cs in the input sequence.
/// </summary>
/// <param name="sequence">The input nucleotide sequence</param>
/// <returns>A sequence of the same length for which each element indicates the
/// count of Gs and Cs between the beginning and corresponding position in
/// the input sequence.</returns>
public static IEnumerable<int> GenerateGCCount(IEnumerable<Nucleotide> sequence)
{
int count = 0;
foreach (Nucleotide n in sequence)
{
if (n == Nucleotide.G || n == Nucleotide.C)
count++;
yield return count;
}
}
Given this sequence (call it s), the density for window size w at some position i could (conceptually) be calculated with the formula d[i] = (s[i+w]-s[i])/w. However, since we might want a large window size, we don’t even necessarily want to have w nucleotides in memory at once, so we can’t use such a simple formula. However, the stream-based version is almost as simple:
/// <summary>
/// Compute the GC density of the specified nucleotide sequence.
/// </summary>
/// <param name="sequence">The sequence of nucleotides</param>
/// <param name="windowSize">Size of the window for each density calculation</param>
/// <returns>A sequence for which each element indicates the ratio of Gs and Cs to
/// total nucleotides over the past windowSize elements</returns>
public static IEnumerable<float> ComputeGCDensity(
this IEnumerable<Nucleotide> sequence,
int windowSize)
{
if (windowSize < 1)
throw new ArgumentOutOfRangeException("windowSize");
// First compute a sequence of the running count of G or C nucleotides
var gcCounts = GenerateGCCount(sequence);
// Combine the sequence with a copy of itself offset by the window size
// The elements of the sequence are pairs of counts, with the first element
// of the pair being the GC count at the beginning of the window, and the
// second element of the pair being the GC count at the end of the window.
var gcWindows = gcCounts.Zip(gcCounts.Skip(windowSize - 1));
// Now for each window, compute the gc density and return it
return gcWindows.Select(
p => (float)(p.Second - p.First) / windowSize);
}
This is pretty much it. If you remove the comments this is only a couple lines of code (or a single long query line if you prefer). Zip is just a helper function I wrote (based on list-processing functions in other functional languages like standard ML’s ListPair.zip or OCaml’s List.Combine) that returns a sequence of pairs from the two input sequences:
/// <summary>
/// A pair (2-tuple)
/// </summary>
/// <typeparam name="T1">Type of the first element</typeparam>
/// <typeparam name="T2">Type of the second element</typeparam>
public struct Tuple<T1,T2>
{
public Tuple(T1 first, T2 second)
{
First = first;
Second = second;
}
public readonly T1 First;
public readonly T2 Second;
}
/// <summary>
/// Combine two sequences into a sequence of pairs
/// </summary>
/// <typeparam name="T1">Type of the elements of the first sequence</typeparam>
/// <typeparam name="T2">Type of the elements of the second sequence</typeparam>
/// <param name="source1">The first sequence</param>
/// <param name="source2">The second sequence</param>
/// <returns>A sequence that is as long as the shorter of source1 and source2.
/// Each element is a pair of values - one from each of the input sequences
/// at the same position. If the input sequences are of unequal length, any extra
/// data in the longer list is not used.</returns>
public static IEnumerable<Tuple<T1, T2>> Zip<T1, T2>(
this IEnumerable<T1> source1,
IEnumerable<T2> source2)
{
// Conceptually we just want to foreach over both sequences, but we must
// write the code manually since foreach works for only a single sequence
var enum1 = source1.GetEnumerator();
var enum2 = source2.GetEnumerator();
while (enum1.MoveNext() && enum2.MoveNext())
{
yield return new Tuple<T1,T2>(enum1.Current, enum2.Current);
}
}
Together I think this is pretty simple and elegant. I create two instances of the same sequence, offset one by the window size, and then use their difference to compute the density. All of this is done on demand each time a density value is required. I’m not keeping any information about any position other than the current one in memory at any one time. One drawback is that for small window sizes, it may actually be pretty silly (from a performance perspective) to read each byte of the sequence from the file twice. But I consider this an optimization problem with the operating system file system cache should ideally solve for me. In practice execution still appears to be CPU bound, so I think the OS is doing a good job – the extra I/O isn’t a concern.
If you’re new to this style of programming, it can sometimes get a little confusing trying to keep track of the timing in which things execute. C#’s iterator method syntax and the IEnumerable pattern in general can make the use of delayed computation a little subtle. This is a big benefit of using a functional programming style like I have where no shared state is modified and the functions have no side-effects. The order in which things execute is unimportant.
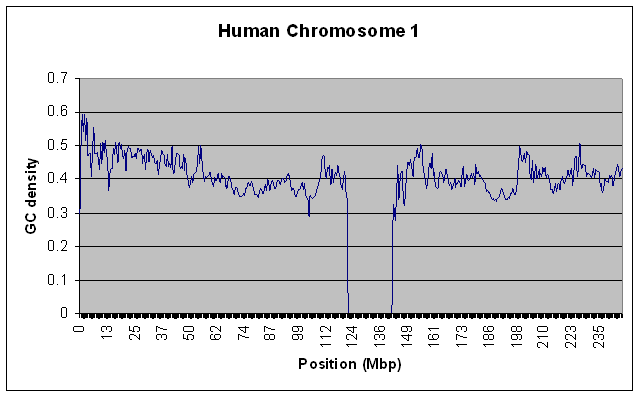
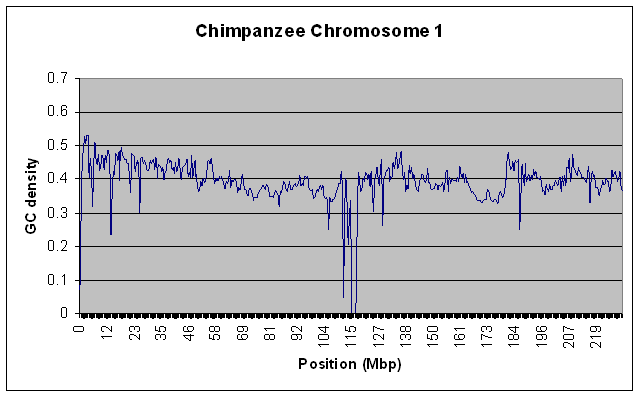
To test that this actually worked on large data sources without using a lot of RAM, I ran it on the entire Human Chromosome 1 sequence (the largest one - almost 300 MB uncompressed) with a window size of 100000. For kicks, I also ran it on the Chimpanzee chromosome 1. Both runs took about 10 minutes on my relatively slow home PC, and used only a steady 10MB of RAM. Here are the resulting graphs if you’re interested.